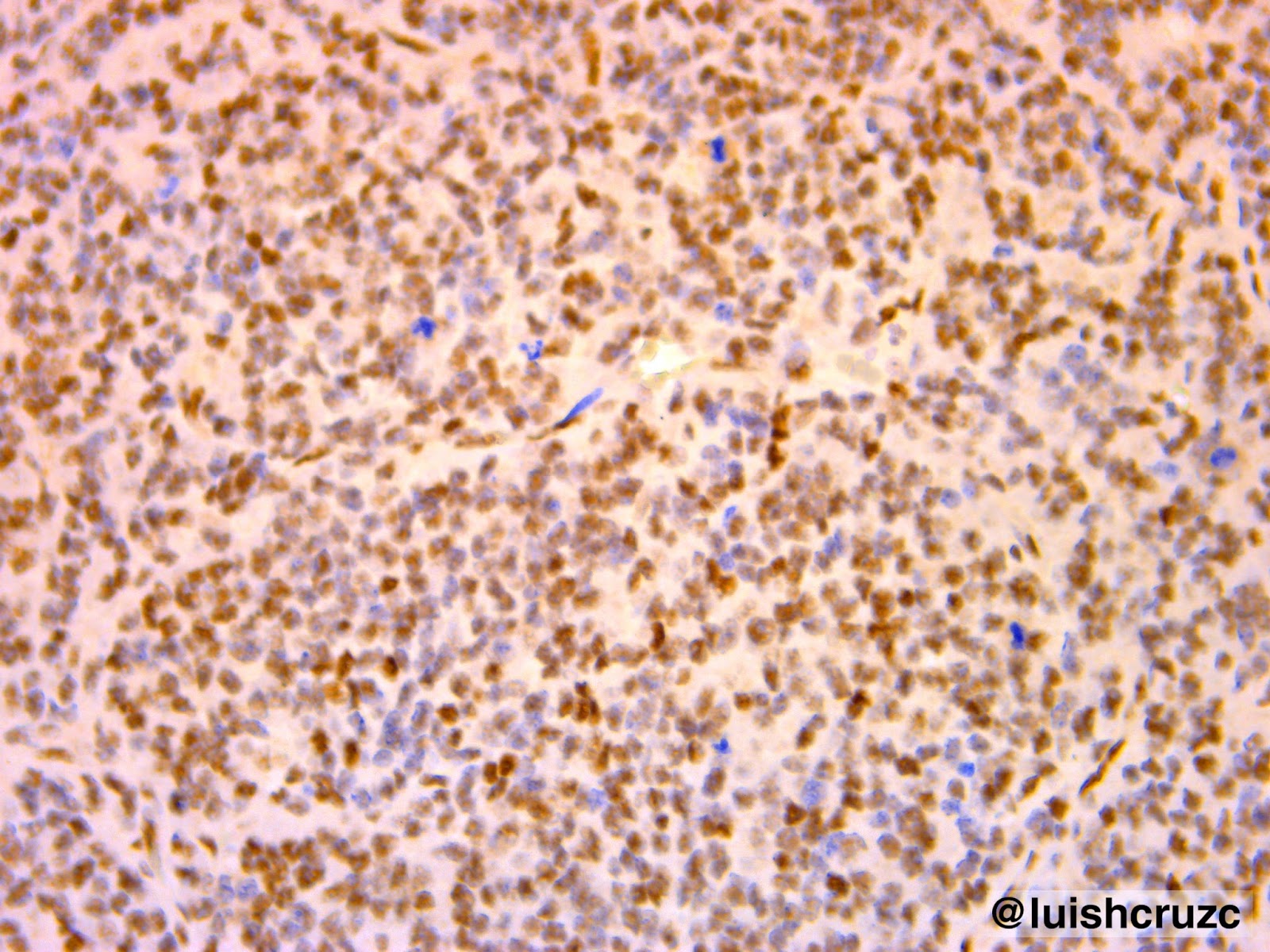
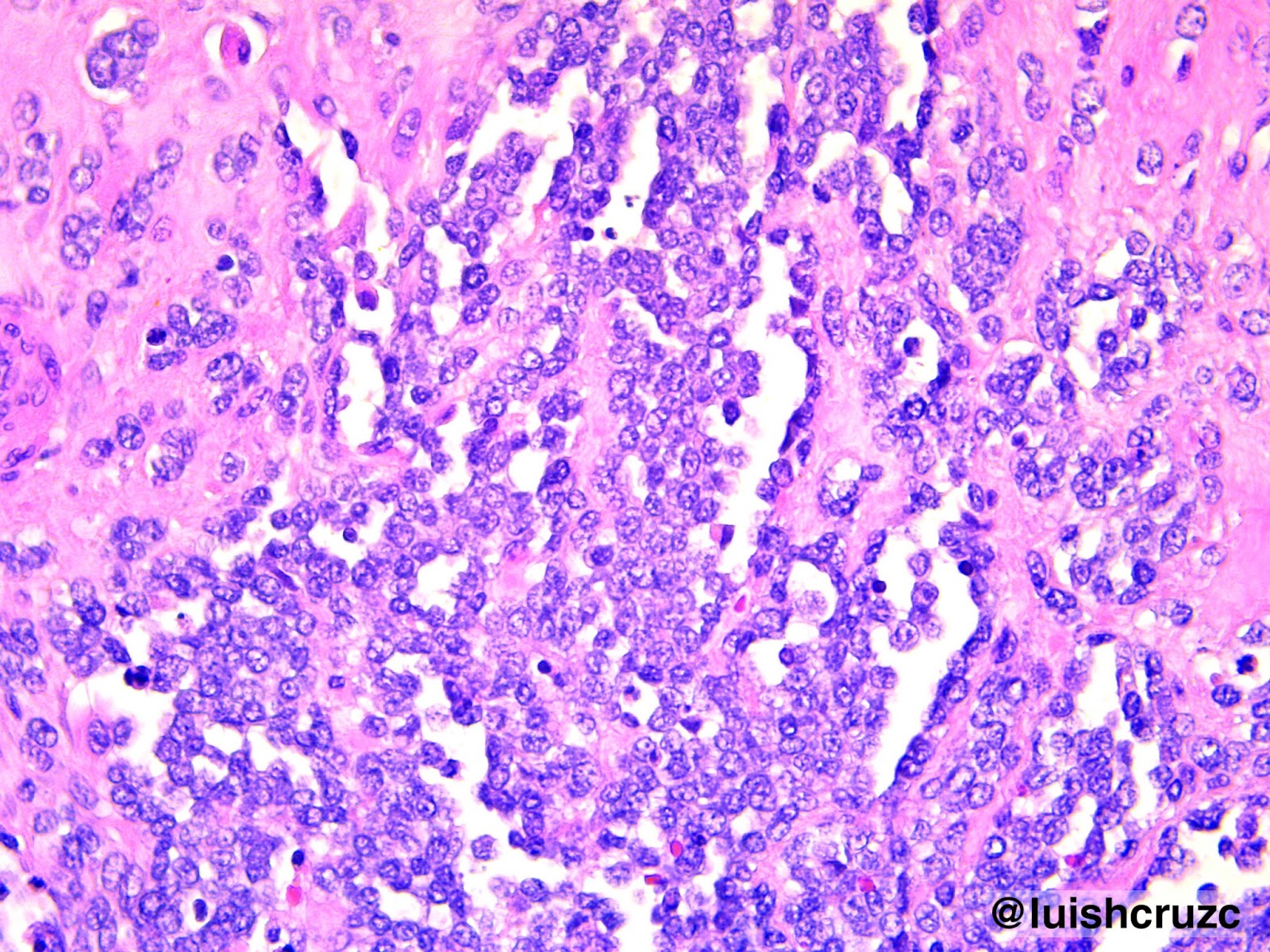
La MCVAP es una lesión pulmonar hamarto-matosa, de carácter congénito y poco frecuente. Representa el 25% de todas las malformaciones congénitas pulmonares, se encuentra en 1 de cada 25.000 a 35.000 embarazos6,8. La MCVAP probablemente resulte de la detención de la maduración bronquial y concomitante sobrecrecimiento de elementos mesenquimáticos. que van produciendo la apariencia adenomatoidea de la anomalía en los estadios precoces del desarrollo, esta área pseudotumoral con tejido pulmonar inmaduro habitualmente recibe circulación pulmonar. A menudo existen pequeñas comunicaciones bronquiales que llevan a la infección e inflamación de los quistes,9-11.
La patogénesis de la MCVAP aún permanece desconocida. Mientras algunos investigadores han formulado la hipótesis de que resultaría de una falla en la interacción entre el endodermo y el mesodermo, otros sugieren un desbalance entre un incremento de la proliferación celular y un detrimento en la muerte celular programada (apoptosis). También se ha considerado que la MCVAP se produciría por una falla en el desarrollo vascular pulmonar, como se ha observado en otras malformaciones12-15.
En 1977 Stocker y cois, basados en la presentación clínica y las características patológicas relacionadas con la madurez del pulmón, desarrollaron una clasificación de la enfermedad16,17. Actualmente, este mismo autor la ha reformado y de tres tipos iniciales ha establecido cinco grupos (tipo 0 a 4) (Tabla 1). Nuestro
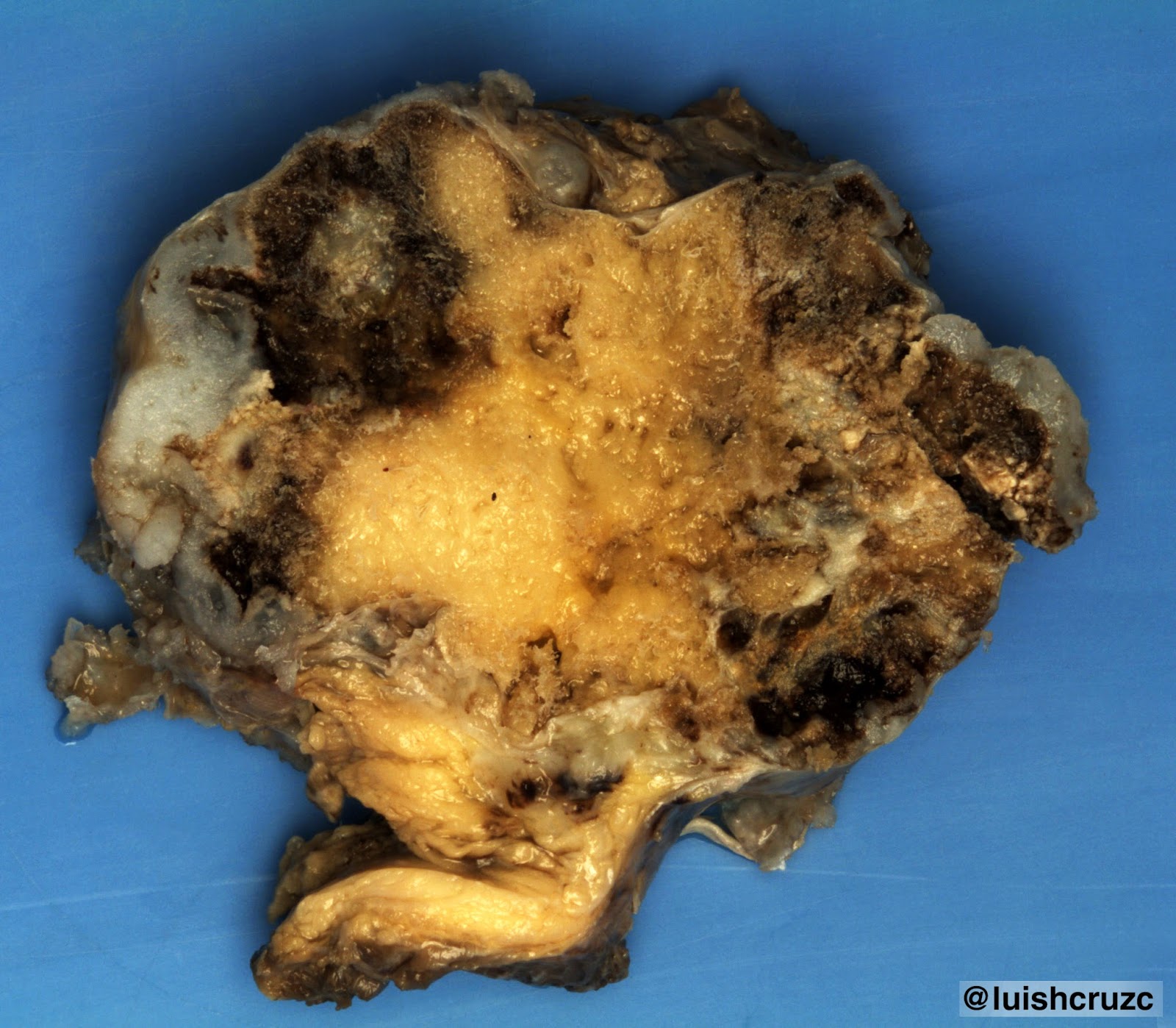
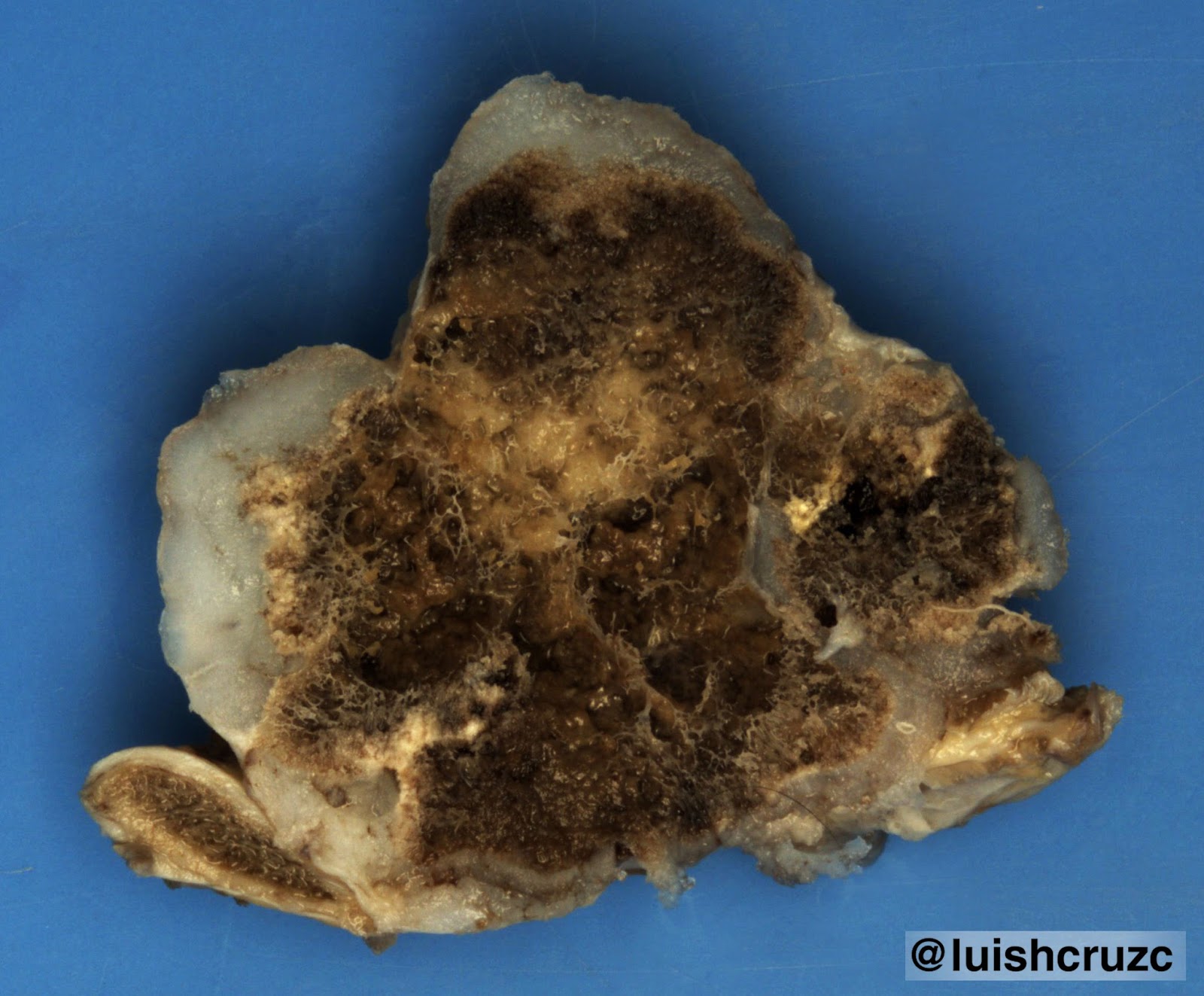
caso corresponde al tipo 1, el más frecuente (> 65%), presentando múltiples quistes pulmonares, el mayor de ellos de 8 cm17,18. La MCVAP generalmente afecta un lóbulo (tal como el caso que reportamos) y cuando es multilobar, sigue siendo unilateral. Todos los lóbulos se comprometen con igual frecuencia. Cuando las lesiones son bilobares o bilaterales, el pronóstico es malo debido a la hipoplasia del pulmón residual2,9-11.
El 83% de los casos se pesquisa antes de los 6 meses de vida. La enfermedad se diagnostica con frecuencia en el período prenatal (18 a 24 semanas), gracias a la ecografía fetal19. Esto permite el tratamiento intrauterino o neonatal inmediato. La sintomatología más frecuente a esta edad es la dificultad respiratoria17,19. A veces está asociado a anasarca fetal, ascitis y polihi-droamnios. No hay predilección por sexo o raza20. La presentación en adultos es excepcional y habitualmente, al igual que en nuestro caso, existen antecedentes de infecciones respiratorias repetidas. No encontramos casos adultos publicados en la literatura nacional. Otras formas de inicio menos frecuentes reportadas en adultos son el neumotorax espontáneo, la bronquitis obstructiva y el dolor torácico. Excepcio-nalmente hay hemoptisis, un micetoma o un absceso pulmonar4,17,19,21,22. Se han reportado casos de transformación maligna en carcinoma bronquioalveolar y rabdomiosarcoma embrionario pulmonar23-27.
[Tomado de:ALVAREZ Z, CARLOS; CERDA C, CÉSAR; CERDA A, CARMEN y SANHUEZA P, BELÉN. Malformación congénita de la vía aérea pulmonar: Reporte de un caso adulto.Rev. chil. enferm. respir. [online]. 2009, vol.25, n.3 [citado 2015-12-15], pp. 182-187 . Disponible en: <http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0717-73482009000300005&lng=es&nrm=iso>. ISSN 0717-7348. http://dx.doi.org/10.4067/S0717-73482009000300005. ]